Blog
New Year, Ready to Restart Your Trial?
Jan 08, 2021 | Natalie Townsend
Jan 08, 2021 | Natalie Townsend
As the COVID-19 pandemic continues to evolve we are all becoming familiar with restrictions easing and expanding both nationally and locally. We are becoming adept at adjusting to the constraints and many sponsors are looking to restart trials that they previously put on hold.
For trials that are restarting, and indeed for all ongoing and new trials, it is important to consider how you will handle and manage the data capture process to account for missing visits, assessments and specific data. In this article we will discuss how to do that.
Key considerations
Ultimately the data must be presented for analysis after the completion of the clinical trial. Both the EMA and FDA have issued guidance highlighting the importance of accurately accounting for all the data that you have been unable to collect, including the reason for why the data is missing.
From the FDAa: ‘’It will be important to capture specific information in the case report form that explains the basis of the missing data, including the relationship to COVID-19 for missing protocol-specified information (e.g., from missed study visits or study discontinuations due to COVID-19)’’.
From the EMAb: ‘’Risk assessment should focus on quality and reliability of the data from a trial conduct perspective and should consider the impact of intercurrent events (e.g. treatment discontinuations) and missing data arising from the COVID-19 pandemic on the analysis and interpretation of the data’’.
It may also be pertinent to consider the design of your protocol and whether any amendments might be necessary, for example you could consider whether to stop collecting data related to an exploratory endpoint. This will help reduce burden on site professionals and study teams as well as risk to patients.
As the situation evolves you will need flexible approaches that allow you to adapt as and when circumstances change.
Potential technology challenges
When defining your strategy for managing missing data and performing mid-study amendments there are technology challenges to navigate. These are nothing new but have been exacerbated by COVID-19.
Firstly, if your EDC solution does not provide a simple mechanism for sites to mark that data is intentionally missing this can have negative effects:
- Sites and CRAs face additional query management burden with back on forth correspondence asking if data is intentionally missing or is genuinely overdue data entry
- Operational reporting on data completeness will be skewed if intentionally blank data is not readily identified
- Study designers may build additional CRFs to capture missing data or add COVID-19 to existing forms and codelists. This workaround is inefficient for data entry as superfluous forms and/or data is introduced for sites to complete. It also adds complexity to data extracts and may require an amendment per study to implement.
Secondly, whilst protocol amendments may be desirable to ease pressure on site personnel and reduce patient risk, traditional EDC systems require data migration and downtime to deliver mid-study amendments. The gains you achieve through your amendment could be compromised by the time, cost, effort and risks to amend your EDC design.
Defining a strategy for success
From a data capture strategy perspective, consider adopting the following practices:
- Focus on the critical data that you need to capture
- Document standard data management guidelines for handling missing data
- Provide sites with a simple method to indicate missing and incomplete data during data entry and the reasons why
- Ensure intentionally left blank data is easily identified as such in the audit trail and in the datasets for statistical analysis
- Adopt a flexible approach to implementing mid-study amendments, an agile design approach will help you to do this rapidly
How can Veeva Vault EDC help?
Using Veeva’s modern cloud based EDC, sites can easily mark data points, forms, and visits as intentionally blank and include the reason for this as part of their natural data entry flow. This provides those reviewing the data with more accurate missing page reports. Additionally, because Veeva can include intentionally left blank data items within the EDC extracts this provides clear visibility into the precise volume of data impacted as per regulatory advice.
A centrally managed drop-down list of reasons for missing data is available to all your studies. The list is easily updated with a simple configuration without the need for individual study amendments or re-validation. Your strategy is future-proofed — allowing you to easily add new reasons should other unforeseen events emerge.
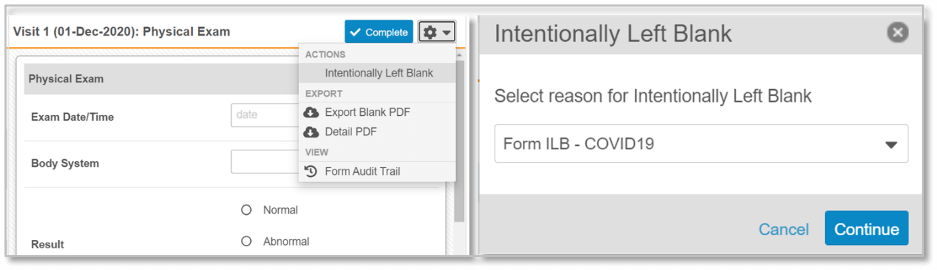
Figure 1. Sites are able to select that an entire event did not occur, or that forms or individual data fields have been left blank intentionally. A configurable drop-down list of reasons why is presented. The designated reason is clearly identifiable during SDV, within CDMS reporting, and in data extracts.
Study amendments in Vault EDC can be rapidly deployed and do not require data migration or downtime. Deployment to specific countries, sites and even to individual patients is easily handled. Therefore if you do choose to update your protocol design, e.g. stop collecting data for exploratory endpoints, you can achieve this amendment in EDC with simplicity and speed.
Veeva Vault EDC equips companies to respond faster to changes and new demands, contact us to learn more.
References
a – FDA Guidance on Conduct of Clinical Trials of Medical Products during COVID-19 Public Health Emergency Guidance for Industry, Investigators, and Institutional Review Boards March 2020
b – EMA, Points to consider on implications of Coronavirus disease 5 (COVID-19) on methodological aspects of ongoing clinical 6 trials