EU MDR and Beyond: The Balancing Act Between Innovation and Compliance
By Annemien Pullen, PhD.
We have entered a new era of continuous global regulatory change. The new Medical Device Regulation (MDR) took effect on May 26, 2021, bringing significant compliance requirements for medical device companies.
Since the European Commission published the final text on 25 May 2017, device manufacturers of all sizes have been working tirelessly to make revisions and mitigate the impact on development pipelines.
However, challenges bring innovation, and forward-thinking companies see MDR as an opportunity to harmonize and modernize processes and technology to gain market advantage.
MDR challenges medtech’s innovation capabilities
The new regulation applies to every medtech company with products on the market in Europe and other regions that apply the CE-mark, such as Japan and Australia. Regardless of company size, the challenges that come with MDR are similar and impact the entire medical device value chain and product lifecycle. Examples of these challenges include:
Disconnected teams and systems
Although some companies are taking strides to enable unified and connected operations, many still run in organizational silos, resulting in disconnected data, processes, and systems. These silos make it difficult, if not impossible, to address changes or inconsistencies quickly.
MDR affects all departments and requires close collaboration across the entire organization. Even the smallest process change calls for Standard Operating Procedure (SOP) revisions, so having just one group drive the change increases the risk of non-compliance. All parts of your business need to be taken into account, including processes, documentation, and systems to be audit-ready and compliant.
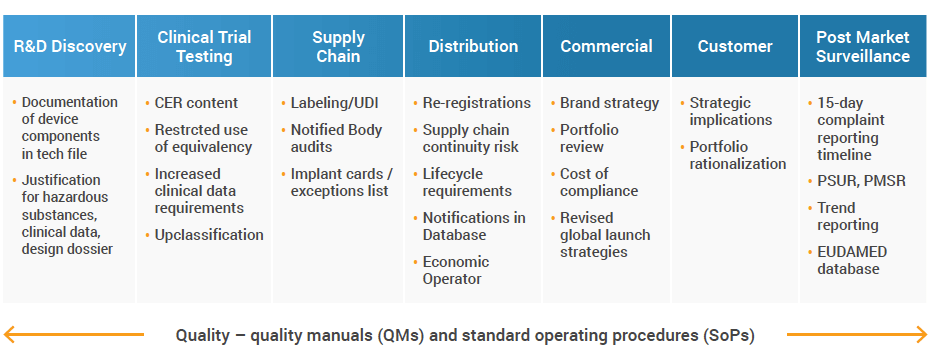
When you look at something like the reporting requirements for the EUDAMED database, you have to pull a significant amount of cross-functional data, while verifying the information and mapping its origins. This alone requires a tremendous amount of time, resources, and data analytics.
Every single department and function need to sustain and maintain MDR compliance. The only way to do that effectively is to work cross-functionally, ensuring that processes are harmonized, conveyance between different departments is clear, and there is one source of data for everyone to access.
Additional data and post-market surveillance
MDR brings additional data and reporting requirements, not only in terms of frequency but also volume. Further to the existing Clinical Evaluation Report (CER), and Adverse Event Reports already present in the Medical Device Directive (MDD), there are brand new ones like Trend Reports (the actual reporting on trends, unlike trending included in the MDD) and Periodic Safety Update Report (PSUR).
These reports all pull data from different business areas, like the benefit-risk analysis, clinical evidence, the main findings of the Post-market Clinical Follow-up (PMCF), and device sales volume with information on the population using the device, or complaints data, to name a few.
And if gathering and consolidating the information wasn’t challenging enough for many businesses, MDR shifts the passive post-approval management to active and ongoing documentation monitoring, update, and renewal. Any change in clinical evidence, claim, risk, or adverse events require reporting and/or submission. For bigger companies, this could lead to 800+ submissions and reports per year.
Maintaining MDR post the DoA
Maintaining MDR post Date of Application (DoA) represents the biggest ongoing challenge for medtech companies. It is fundamental to establish sustainable MDR continuation now to achieve long-term business continuity and compliance.
To meet the requirements many organizations initiated MDR programs and have most of the certificates in place. However, a program is a temporary structure as it only focuses on meeting the initial submission compliance requirements. What happens when these programs end? Who is responsible for managing the ongoing MDR requirements?
In some companies, handover discussions are happening right now. However, not all yet recognize the need to reorganize operating models and put appropriate tools and technology in place in parallel with the program to set up long term success and sustainability.
These are just a few factors that will challenge medtech companies, causing significant increases in costs, possible reduction in a product’s revenue potential and less room for investments in R&D.
It is estimated that the MDR transition can cost device manufacturers between 3.5% and 5% of total revenue in Europe.6 Companies with large and profitable product portfolios may afford additional investments, but smaller companies do not have the same luxury. Instead, they may see decreases in the development pipeline, as they shift resources to support compliance.
According to Peter Gaines, professor at Sheffield Hallam University, approximately 50% of all medical devices will be withdrawn from the market; bigger device manufacturers will halve their product portfolio, and 30% of smaller manufacturers will simply shut down.1

MDR drives operational modernization
As said, MDR affects all departments and requires close collaboration across the entire organization.
MDR is not a do it once and forget it initiative. It requires continuous maintenance and scrutiny; the Technical Documentation and broad variety of regulatory reports are not static but is a consolidation of dynamic information that evolves as the product and its use in the market evolves. For example, post-market data and additional clinical evidence might result in new or revised claims that impact the technical documentation and associated reports.
As the businesses take over responsibility from the MDR program to maintain the MDR post-DoA, workarounds and adding people can quickly become unsustainable. To achieve long-term continuity and compliance the business must implement sustainable solutions to operating models, governance structures, and systems.
That’s why the traditional approach of filling gaps in the existing processes or developing workarounds to maintain compliance will no longer work. Of course, given the limited time and budget and the need for business continuity, many companies have taken a short-term focused approach to MDR.
However, it is not sustainable to maintain MDR in this way in the long-term. A sustainable MDR operating model requires medtech companies to:
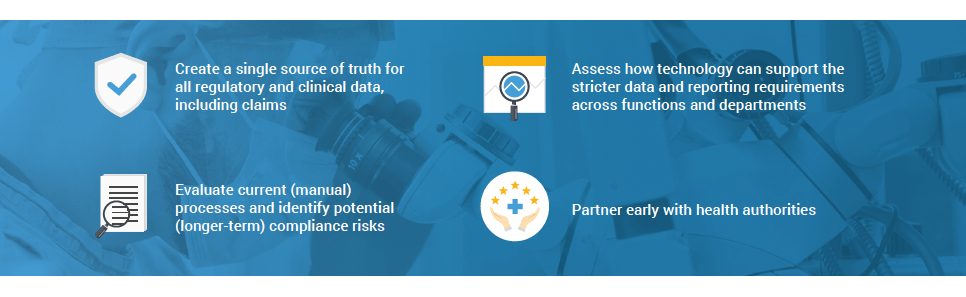
Reshaping the business with technology
The MDR is not meant to be easy. However, forward-thinking companies are innovating and embracing digital technologies to unify operations and ensure compliance.
Organizations like Alcon and GE Healthcare have put Regulatory Information Management (RIM) Systems in place to build their submissions, track and manage their global product registrations, and handle their healthcare correspondence.
They can manage the end to end regulatory process throughout the product lifecycle and know the status of their business 24/7 and without manual interference. This allows their highly skilled regulatory professionals to focus on regulatory strategy and other value-adding activities rather than administrative tasks.
These companies are getting ahead of new regulations by facilitating better access, visibility, and control over documents and data across the company.
Digital transformation: unified and connected medtech
Alcon, a leading ophthalmology devices manufacturer, replaced outdated, siloed systems with a unified and connected cloud platform to manage all regulatory content and data from one system.
Before this, Alcon handled submissions and registrations manually, using spreadsheets and email. If Alcon had a technical document that supported five technical files, they had to have five versions of that technical file in the system.
Now they have a single source of truth for all regulatory data. They’ve eliminated duplicate content and have reduced time to get approvals with automated workflows.
Lori Holder, director of global regulatory operations at Alcon, remarks:
“With our unified approach, all that data is contained in one system. We can take that information and upload templates, request funding, and completely manage submissions without emailing back and forth. We are also able to collaborate on final drafts of submissions and submit them for approval electronically.”7
Similarly, GE Healthcare, a leading global medical technology and diagnostics company, uses a unified RIM solution to automate tasks, reduce administrative burden, and free up valuable resources for more value-add tasks. The company and their affiliates can now easily access one system to update data, removing manual methods like filling out a form and emailing HQ for data entry.8
As a result, GE Healthcare cut down the time required to create records for a regulatory activity across 130 markets from several hours to just five minutes.9
Digital technologies enable companies to optimize and harmonize the end-to-end processes. They accelerate efficiency and unify operations, which allows the highly skilled staff to focus on strategy and other value-adding activities. In the long run, this allows these companies to get ahead of ongoing global regulatory change and bring their products to market faster. Finally, it is key for business continuity when faced with any forthcoming changes like MDR.
By Annemien Pullen, PhD., Director Strategy, Veeva MedTech Europe
References
2 Howard T., 2020. When “Business as Usual” Falls Short [online] veeva.com
3 Keogh, S., 2012. Poly Implant Prothèse (PIP) Breast Implants: Final Report [online] gov.uk
4 Roderick, L., 2017. The fight for women poisoned by breast implants [online] theguardian.com
5 Keutzer, L., Simonsson, U., 2020. Medical Device Apps: An Introduction to Regulatory Affairs for Developers [online] mhealth.jmir.org
6 Busscher, L., Flockhart, A., Baylor-Henry, M., Brennan, J., Giovannetti, G., Kumli, F.,2017. How the new EU MDR will disrupt and transform the industry [online] eyjapan.jp
7 Howard, T.,2020. How the Role of Regulatory Operations Professionals Will Evolve [online] veeva.com
8 Howard, T., Scogland, B., 2020. Four Key Areas To Streamline Regulatory Operations and Compliance [online] veeva.com
9 2020. GE Healthcare Streamlines Global Regulatory Processes with Veeva Vault RIM [online] veeva.com
When connecting QA and RA, medtech leaders deliver higher quality products to market faster while improving their bottom line. Read more in our new whitepaper Harmonizing Quality and Regulatory to Streamline and Speed MedTech Product Lifecycle.