How to Tackle Complex Medtech Regulations with Existing Resources
As the demand for new technology grows, the pipeline for medical devices and diagnostics is expected to increase at a compound annual growth rate of 10% through the year 2030.1 Further accelerating this growth is an explosion in enabling technologies like AI, robotics, and 3D printing plus new sustainability and net zero requirements. All of these factors affect the way that devices and diagnostics are manufactured and regulated.
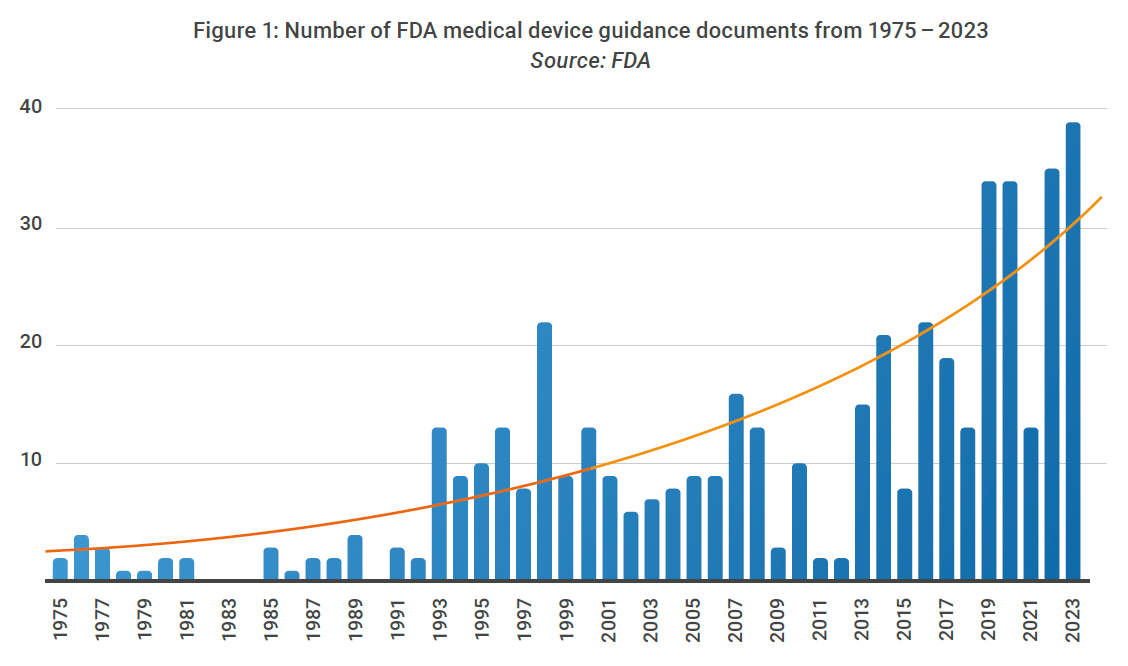
In response, global health authorities are issuing new regulations at an exponentially fast pace. Between 2015 and 2022, there was a 64% increase in regulations for medical device manufacturers, reaching over 13,000 regulations in 2022.2 As shown in Figure 1, there were more FDA guidance releases in the past six years than there were in the previous 40+ years.3
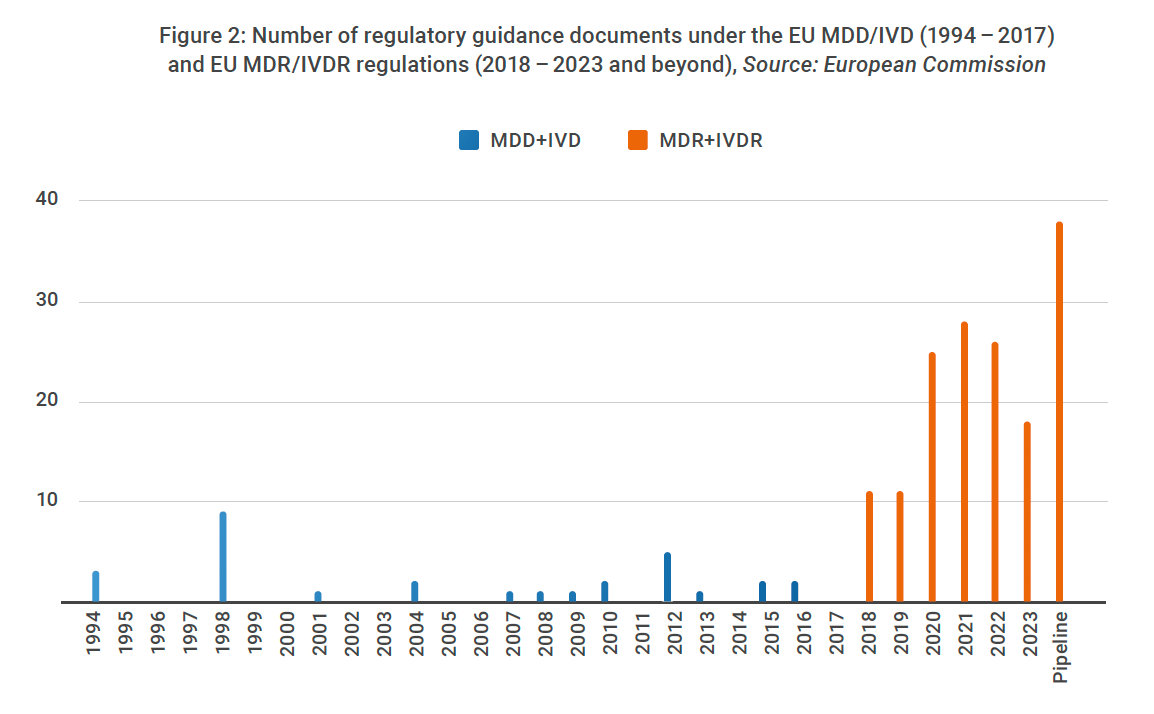
Numbers in the EU tell a similar story. As seen in Figure 2, there were only 30 regulatory guidance releases between 1994 and 2017 when MDD and IVD were in place. But that number leaped to 124 between 2017 and 2023 with the emergence of MDR and IVDR.4,5
Increasing regulatory activity means greater challenges
This surge in regulatory activity creates a very difficult environment for medtech organizations that are trying to bring new products to market while maintaining compliance for their existing portfolios of devices and diagnostics. The brunt of that burden falls on regulatory affairs (RA) professionals, who grapple with multiple competing priorities:
- Managing ambiguity: As shown in Figures 1 and 2, regulatory guidance is issued over the course of many years, leaving RA teams to make decisions based on partial information. This ambiguity can be incredibly frustrating and results in delays and lost revenue opportunities when plans need to shift unexpectedly. Therefore, it’s critical for industry stakeholders to monitor and contribute to draft guidance with ‘real-life’ concerns and examples so regulators understand the impacts.
- Dealing with bottlenecks: When it comes to new product development, RA is inundated with data including product information, registration information, submission dossiers, and marketing plans. In most cases, this information is siloed in different systems, subject to duplication errors, and assembled manually through slow, outdated processes. In a recent Veeva regulatory benchmark survey, a mere 14% of study participants said they have a single-source-of-truth platform to exchange strategic plans across functions. This creates countless bottlenecks and slows down go-to-market timelines.
- Resubmitting information multiple times: After a submission is complete, there’s no guarantee that it will be accepted. In fact, 20% of 510k submissions are not accepted for initial review by the FDA and two-thirds require additional information.6 In the EU, about 25% of MDR applications are refused.7 The reasons can range from incompleteness to incorrect classifications or qualifications, creating a vicious cycle of reviews and resubmissions. RA teams will often need to reallocate resources and put other projects on hold until the submission is approved.
Tackling higher costs with limited resources
The costs associated with obtaining market approval are also at an all-time high. The amount of total company funding to develop a Class II 510(k) cleared medical device is approximately $30 million, with $24 million of that spent on FDA-related activities.8 If your company is working on a high-risk Class III device, that total number skyrockets to $75 million.
RA teams are also struggling to find new talent, which may only get worse as projections estimate 70,000 regulatory job openings over the next decade.9 This means RA teams must learn how to handle greater volumes with the same number of employees.
Leveraging technology to keep up with new regulations
Despite all these negative factors at play, RA is ideally positioned to help drive innovation and empower teams to bring new products to market faster. However, to fulfill that strategic role, RA professionals need more technology to scale their efforts and keep up with regulatory demands. Many companies are now turning to regulatory information management (RIM) systems to centralize information, streamline operations, and accelerate regulatory timelines while maintaining compliance. These solutions can support the full regulatory lifecycle and help answer questions like where products are registered and when they’re due for renewal. For a comprehensive overview on scoping and selecting a RIM solution, read this guide.
By utilizing a single source of truth to store, validate, and access information, RA teams can save time while enhancing visibility and control over global registrations. For example, GE Healthcare saved 200+ hours annually by centralizing product approval and renewal reporting in Veeva RIM. The company also cut the time it takes to update country records by 95%, bringing a two-hour-long process down to just five minutes.10
Veeva RIM doesn’t just help companies manage product registrations, it also provides a unified platform for submission authoring, publishing, and archival. With this solution in place, a global diagnostics leader reduced their timelines for submitting technical documentation to new markets down to as little as two days. This level of efficiency allows medtech companies to function as truly global regulatory organizations with visibility across teams and regions.11
Learn more and connect
To learn more about selecting the right RIM solution for your team, read this buyer’s guide.
References
1. Grandview Research, Medical Device Regulatory Affairs Market Size, Share & Trends Analysis Report, 2022↩2. Compliance & Risks, Regulatory Trends in Medical Devices, 2023↩
3. FDA, Guidance Documents (Medical Devices and Radiation-Emitting Products), 2024↩
4. European Commission, Guidance MEDDEVs, 2022↩
5. European Commission, Guidance – MDCG endorsed documents and other guidance, 2024↩
6. FDA, 4th Quarter 2023 MDUFA V Performance Report, 2023↩
7. European Commission, Notified Bodies Survey on certifications and applications (MDR/IVDR), 2023↩
8. StarFish Medical, How Much Does it Cost to Develop a Medical Device?, 2023↩
9. Research Features, Talent shortage in regulatory affairs is cause for concern, 2021↩
10. Veeva Systems, GE Healthcare Streamlines Global Regulatory Processes with Veeva RIM, 2020↩
11. Veeva Systems, Roche Diagnostics and Veeva MedTech, 2021↩