White Paper
Despite EMA’s Agile Delivery, There Is Limited Breathing Space for IDMP Preparedness
By focusing on their data, processes, and organizational models, sponsors can accelerate regulatory transformation.
Following the delivery of some key elements in 2023, EMA’s Identification of Medicinal Products (IDMP) mandate continues to absorb considerable effort. The industry has welcomed some of the changes underway: this includes transforming electronic application forms (eAFs) from cumbersome PDFs to web-based forms and integrating them with the product database on the product lifecycle management (PLM) portal. Providing the appropriate eAFs via the PLM portal will reduce the burden on companies to create custom data packages for IDMP.
There are also signs of greater flexibility in EMA’s delivery model and sponsor engagement. ‘Big bang’ implementation, which would have meant getting everything right on day one, has evolved into an approach of pilot, learn, and adapt. This alleviates the pressure on sponsors, particularly as the structured product data scope expands this year from 15 structured data fields to eventually enable the full “iteration 1” scope, as defined in the latest implementation guide.
However, a phased implementation offers limited breathing space. Given the U.S. Food and Drug Administration and Swissmedic are working to find common ground with EMA, IDMP will become the global data standard for key product information. This requires a scalable approach to data, processes, and governance across companies’ global footprints.
Building a base for continuous transformation
Based on the current timescales, it will be mandatory to use the new web-based eAFs for post-approval variations by the end of 2024. One of the key initial activities will be to assess the accuracy of product data currently held by EMA for both centrally and nationally authorized products. Companies will be asked to correct and/or enrich their datasets using Product User Interface (currently in development by EMA and available to the industry in 2024).
Subsequently, other types of procedures (such as initial marketing applications and renewals) will be enabled by the eAFs. PMS product data will also be used by other EMA projects, such as the European Shortages Monitoring Platform (ESMP) and Antimicrobial Sales & Use (ASU). In Q4, it will become mandatory for the industry to update and enrich manufacturer and packaging data via these mechanisms. The ‘critical medicines’ category will be prioritized to pre-empt the risks of drug shortages during crises.
In regulatory terms, the end of the year is just around the corner. Companies that have not started preparing will soon encounter difficulties with data readiness, data management processes, and organization. These initiatives can take between 12 to 24 months to embed properly.
IDMP data maintenance prior to the actual submission requirement can be resource-intensive and costly. For this reason, many smaller companies have been reluctant to start inputting data into their systems. Larger companies similarly found it challenging to plan, given the uncertainty around resourcing. However, regulatory teams that started early are experiencing tangible benefits, including viewing data earlier because they have stronger connections with the right stakeholders.
By regulating data intake, processing, and reuse, IDMP standards are widely expected to usher in continuous transformation, helping life sciences to become more responsive to the patient community. Digital forms are only the tip of the iceberg. Use cases such as predicting and preventing drug shortages were not foreseen six years ago when the principal objective was pharmacovigilance.
Reinforcing data control and ownership
How should companies prioritize? Over the last 12 months, sponsors tracking their progress with PLM have gradually implemented changes. Identifying and validating all relevant internal data for IDMP is foundational work yet challenging. This is because not all data is stored in databases, so regulatory teams locate data points manually in spreadsheets and documents, including the summary of product characteristics (SmPC) and other text formats.
Once found, this data must be tracked at the first point of capture and stored securely in the right format and place. Legacy RIM repositories are not well-suited to be a single source of truth, leaving some companies to manually patch up their data before it can be sent to EMA. Given the genesis of much regulatory information from outside regulatory (for example, in clinical, product supply, or pharmacovigilance), this involves significant cross-functional coordination. In other instances, it’s been time-consuming to create structured formats from information that used to be copied and pasted when shared between departments.
With an estimated increase of up to 60% more data involved than in xEVMPD, IDMP also poses a serious challenge to data control and ownership. Leading companies are now focused on establishing a solid data governance strategy, which includes aligning data management processes with IDMP expectations, assigning data owners and stewards, establishing clear expectations, roles, and responsibilities, and defining unambiguous data standards. Fortunately, IDMP’s agile delivery has empowered them to test, learn, and adapt.
Jazz Pharmaceuticals, for example, realized the need to standardize data entry and decided to change its internal resourcing: central RIM teams are now responsible for creating registrations, while decentralized teams manage data entry. As Gareth Markham, associate director for regulatory information management, explains, “Central teams understand the system requirements; decentralized teams know the data, can ensure accuracy, and are more scalable as we enter new markets.”
Choosing once, choosing right
Once they have streamlined their cross-functional data processes and planned effective resourcing, sponsors need to decide on their technology approach. This can be a complex decision because the regulatory agenda is both dynamic and dense. Organizations are unable to see around the corner, which can lead to inefficiency if they decide to introduce a new information management system for each regulation [Figure 1].
For instance, although it will be replaced by IDMP, xEVMPD will remain the core of all Eudravigilance-related systems at EMA for the foreseeable future and is still the only means of updating authorized product data across all types of procedures. IDMP will replace xEVMPD for data updates in a stepwise approach (and even once xEVMPD data updates are turned off, it will continue to provide support to Eudravigilance systems).
Figure 1: A dense regulatory agenda for sponsors
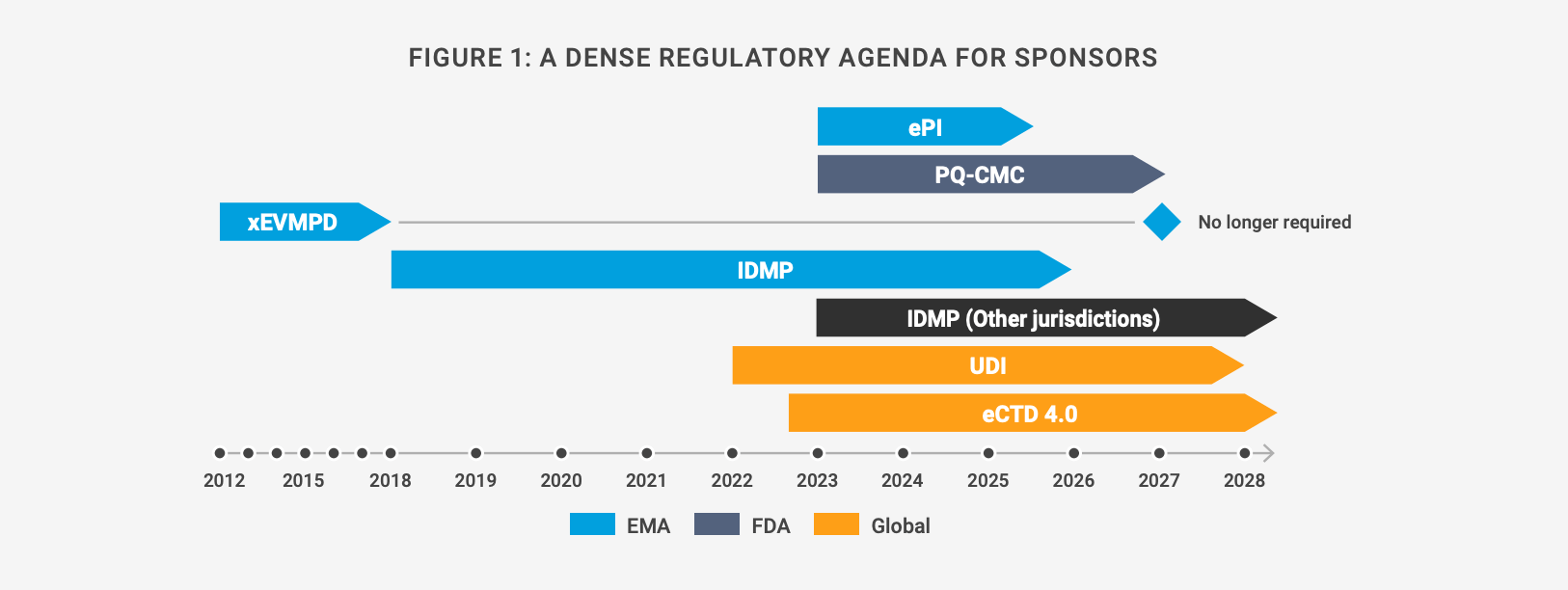
Approaching each regulation with a point solution isn’t viable, given the volume of data, documents, and content covered by the rules, and evolving requirements for authorizations at different levels [Figure 2]. Treating IDMP as an add-on to the core RIM system would also incur additional resourcing, costs, and technology footprint. Several solutions would be needed to account for local submissions in all countries, for instance.
Figure 2: Point solutions are unlikely to work
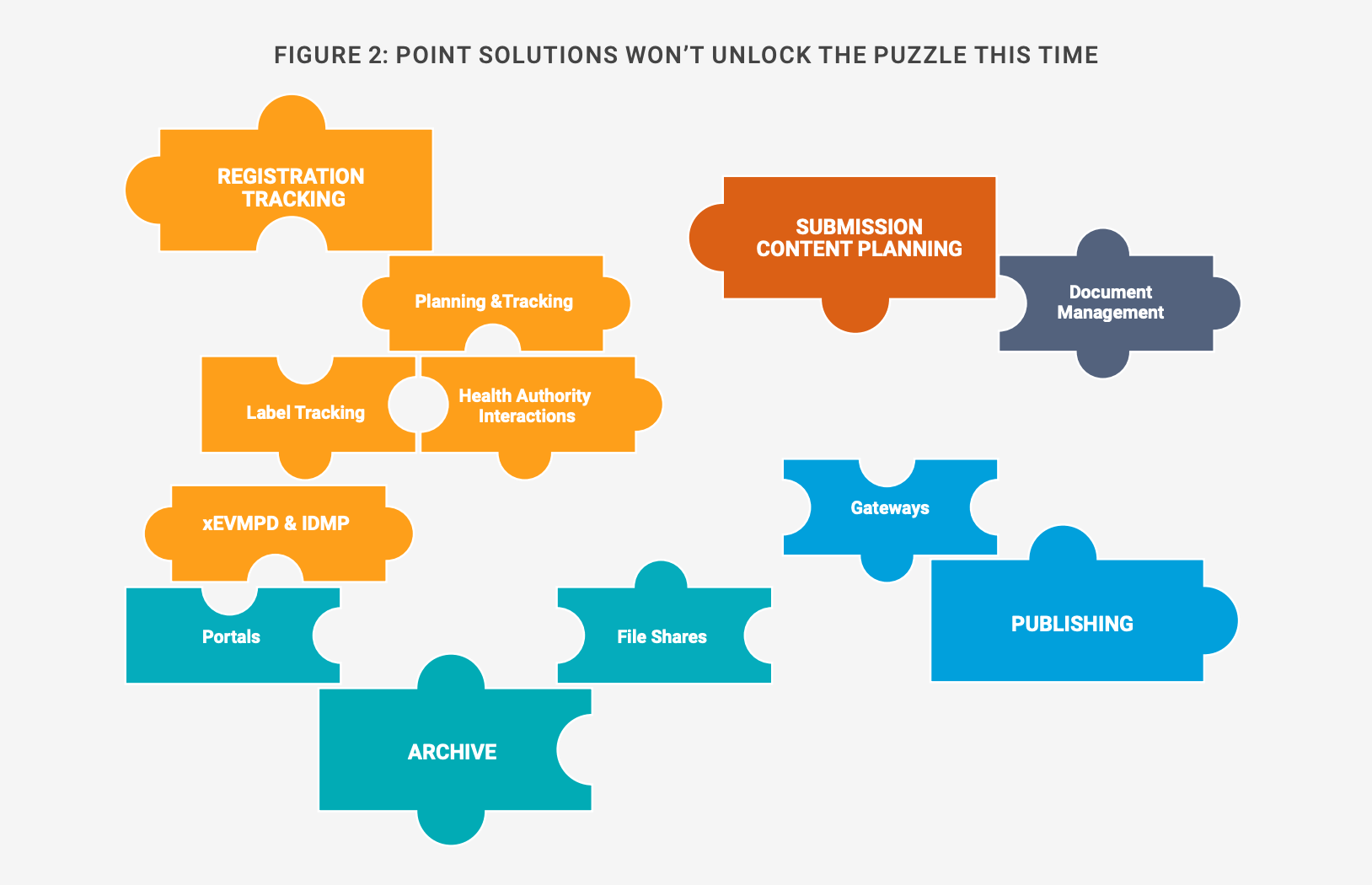
A unified RIM foundation will prove more effective than point solutions, including during the transition period between xEVMPD and IDMP. Although there are key differences between the two regulations and the data sets underpinning them, the core information is the same, which makes it straightforward to set up a single data governance model.
When all core data for IDMP can be sourced from the same system being used for regulatory submissions, teams can share responsibilities without blurring accountability (for example, when sending data to the regulator). Better accountability drives higher-quality metrics. Over time, we expect the single data governance model to be adaptable to more decentralized data sets, like the FDA’s upcoming PQ/CMC.
Solving a complex jigsaw puzzle
By instituting a structure where previously there was none, IDMP will streamline the health information exchange between EMA and the FDA. Regulatory authorities will gain more useful insights than possible through documents alone for improved safety, drug supply management, and public health.
Laggards to digital transformation in regulatory will find that IDMP drives their organizations to maturity quicker than anticipated. Leading companies — which already have a good handle on data quality, ownership, and governance — won’t approach IDMP purely as a regulatory challenge and will focus instead on embedding effective master data management so they can drive business benefits beyond compliance within their organizations.
Biopharma companies are attempting to solve a complex regulatory jigsaw puzzle by piecing together data, documents, technology, and processes to meet the new global standard. Without a doubt, it’s a multi-phased challenge. But a life sciences sector that delivers better patient health outcomes is a prize worth pursuing.
Register to attend Veeva’s R&D and Quality Summit in Madrid to learn more about IDMP readiness.